
An der Bekämpfung von Krankheiten durch Medikamente sind viele unterschiedliche Moleküle im menschlichen Körper beteiligt. Doch meistens beeinflussen nur wenige den Krankheitsverlauf signifikant. Diese seltenen Bausteine gilt es bei der Entwicklung neuer medizinischer Wirkstoffe zu finden, was der Suche nach einer Nadel im Heuhaufen ähnelt. Ganze 13 Jahre dauert es im Durchschnitt, bis ein Arzneimittel fertig entwickelt ist. Um diesen Prozess zu beschleunigen, arbeiten Novartis und Microsoft an Techniken zur Entdeckung neuer Wirkstoffe durch künstliche Intelligenz (KI).
In der Vergangenheit beruhte das Finden von Wirkstoffen oft auf dem sogenannten Serendipitätsprinzip, bei dem glückliche Zufälle und intelligente Schlussfolgerungen zu unerwarteten Entdeckungen führen. So führte zum Beispiel eine vergessene Petrischale dazu, dass der schottische Bakteriologe Alexander Fleming 1928 das erste Antibiotikum Penicillin entwickelte, das bis heute erfolgreich eingesetzt wird, um bakterielle Infektionen zu bekämpfen.
Zentraler Bestandteil jedes Arzneimittels ist der Wirkstoff: also jene Molekülverbindung, die im Körper wirklich eine heilende oder lindernde Wirkung erzielt. Bei einer geschätzten Anzahl von einer Dezillion (1060) organischen Molekülen, die man testen könnte, wird schnell klar, wie schwer es ist, gezielt die Verbindung zu identifizieren, die den Krankheitsverlauf positiv beeinflusst. Und wie groß ist überhaupt eine Dezillion? Es wären genug Moleküle, um die Erde milliardenfach zu reproduzieren. Von wegen „Nadel im Heuhaufen“! Das sind noch ganz andere Dimensionen.
Moderne Arzneimittel sind Hightech-Produkte
Aus ihren zufälligen Anfängen hat sich die pharmazeutische Industrie zu einem der technisch fortschrittlichsten Sektoren entwickelt – angetrieben durch Fortschritte in der Chemie und Molekularbiologie. Dennoch wird immer noch viel ausprobiert und experimentiert, auch wenn sich die Entdeckung von Arzneimitteln weit vom Zufallsprinzip entfernt hat. Zunehmend liegt ein rationaler und vielstufiger Prozess zugrunde, der immer mehr von Computer-Technologien begleitet und gestaltet wird.
MoLeR: Mit maschinellem Lernen zu effizienterem Wirkstoffdesign
So kommt Technologie zum Beispiel in der sogenannten Lead-Optimierung zum Einsatz. In dieser Phase der Arzneimittelentwicklung werden Wirkstoffe im Labor überprüft, die zuvor als geeignete Kandidaten eingestuft wurden. Bei der Lead-Optimierung arbeiten erfahrene medizinische Chemiker*innen an der Verbesserung der Verbindungen, die in einem frühen Screening vielversprechende, aber auch unerwünschte Eigenschaften aufweisen.
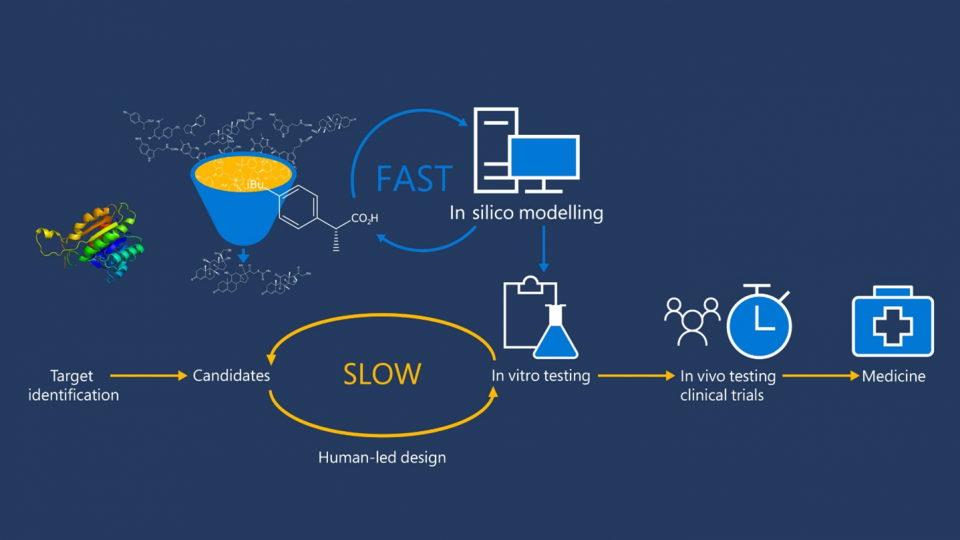
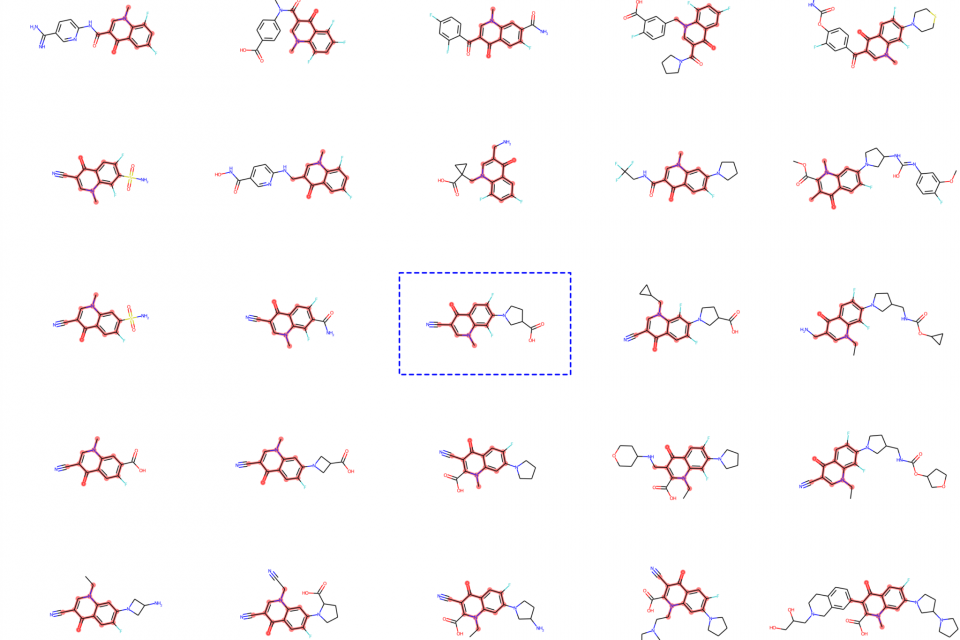
Über die Tests versuchen die Chemiker*innen, die Struktur der Moleküle so anzupassen, dass sich ihre biologische Wirksamkeit verbessert, während zugleich die Wahrscheinlichkeit von Nebenwirkungen verringert wird. Im klassischen „human-led drug design“ ist die Lead-Optimierung ein iterativer Prozess, bei dem neue Verbindungen durch die Forschenden vorgeschlagen und in vitro getestet werden. Da dieser Prozess eine Synthese im Labor erfordert, ist er sehr kostspielig und zeitaufwändig und benötigt darüber hinaus das Wissen, die Kreativität, Erfahrung und Intuition der Forschenden.
Um diesen Prozess der Lead-Optimierung zu unterstützen, wurden computergestützte Modellierungstechniken entwickelt, mit deren Hilfe sich vorhersagen lässt, wie sich die Moleküle im Labor verhalten werden. Durch den Einsatz von Computermodellen kann der Entwurf von neuen Molekülen schnell „in silico“ durchgeführt werden: Alle Modelle werden zunächst im Computer simuliert, und nur die vielversprechendsten Moleküle werden dann zur Herstellung im Labor und schließlich zur Prüfung in vivo vorgeschlagen. So können sich die kostspieligen und zeitaufwändigen Experimente auf die vielversprechendsten Verbindungen konzentrieren, die die beste Wirksamkeit und zugleich die geringsten Nebenwirkungen versprechen.

Um diese Modellierungstechniken zu verbessern, arbeitet unser Generative Chemistry-Team bei Microsoft mit Novartis zusammen. Gemeinsam haben wir das MoLeR-Modell entwickelt: ein neues graphenbasiertes neuronales Netzwerk für maschinelles Lernen. Es ermöglicht die automatische Konstruktion von wirksamen Molekülen in silico und macht die Entdeckung von Medikamenten damit schneller und effizienter.
Wie genau das Forschungsteam bei der Erstellung und dem Training des MoLeR-Modells vorgegangen ist, lesen Sie in dem Artikel „MoLeR: Creating a path to more efficient drug design“ im Microsoft Research Blog. Die wissenschaftliche Publikation „Learning to Extend Molecular Scaffolds with Structural Motifs“ sowie den über GitHub verfügbare Trainings- und Inferenzcode für das MoLeR-Modell finden Sie hier.
Ein Beitrag von Melanie Weber
Industry Executive – Chemical, Pharma & Life Science Industry



